|
Dr. Jan O. Korbel |
|
Computational Biology & Genomics, Gerstein lab |


|
Protein Function Prediction |
|
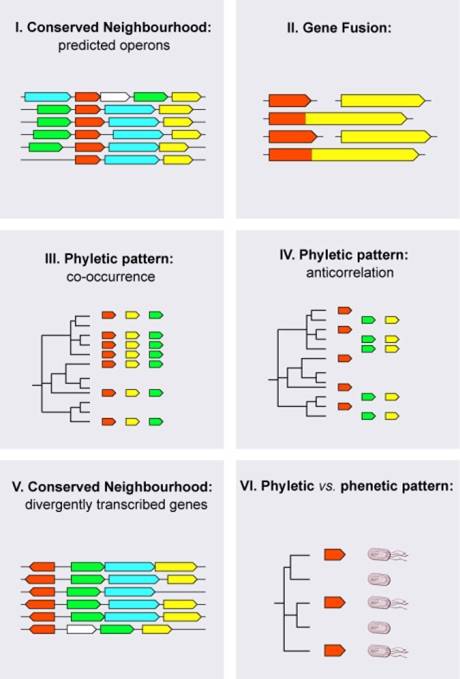
Many genome sequences have been completed over the last years and the pace of DNA sequencing is constantly increasing. When analyzing a newly sequenced genome it is typically impossible to assign functions to more than two thirds of all protein-coding genes, even if experimental approaches for function characterization as well as computational algorithms for homology detection (for example, BLAST [1]) are applied extensively. There is thus a great need for reliable tools for protein function prediction. A recently developed class of analysis approaches, the genomic context approaches, is often well-suited for function prediction, and can be utilized to infer functional aspects overlooked by BLAST. Genomic context approaches analyze in detail the genome a protein is encoded in: the genomic ‘background’, or context, points at the protein’s cellular role. While I was a graduate student at the EMBL Heidelberg in the laboratory of Peer Bork, we both used previously existing genomic-context based methodologies for function prediction (i.e. Approaches I-III), and together with collaborators developed three novel genomic-context approaches (i.e. Approaches IV-VI): Approach I: Conserved neighborhood, predicted operons. For example, prokaryotic proteins which participate in a common biochemical pathway or cellular process are often encoded by neighboring genes in the genome. Together, the genes form an operon, a group of genes that can be regulated and translated into proteins simultaneously. The ‘conserved operon approach’ (for illustrations, see Figure 1below) looks for operons in many completely sequenced genomes. If genes form a similar operon-structure over and over again (in this case the operon is presumably highly evolutionarily conserved), it is very likely that the respective proteins are functionally associated [2,3]: in other words, they are members of the same biochemical pathway or cellular process, or they form a protein complex together. Approach II: Gene fusion. This approach searches for large fusions of protein-coding genes. If the open reading frames (ORFs) of two distinct genes frequently appear merged in different genomes, the two polypeptide halves of the resulting large fusion protein are likely to be functionally associated [4,5]. Moreover, also the corresponding, separately occurring proteins in other species are in such a case likely to interact. Approach III: Phyletic pattern, co-occurrence. This approach uses the fact that groups of proteins involved in certain cellular pathways or complexes are usually together conserved in a species, if the pathway or complex is present in the organism [6,7]. In other words, the corresponding genes are usually either together present or absent from genomes (i.e. they co-occur).
Approach IV: Phyletic pattern, anticorrelation. If the presence of two genes across eukaryotic and prokaryotic genomes is mutually exclusive – in other words the gene occurrences 'anticorrelate' (one gene is present in a genome and the other one is absent) – it can be assumed that both genes encode proteins with the same function [8,9]. The underlying rationale is that there is usually no need for a genome to encode the same function twice. We have developed an approach that exploits gene occurrence anticorrelation in order to systematically identify functionally equivalent proteins [10]. We applied it for the discovery of analogous enzymes in the thiamin biosynthesis pathway . If catalytic function and cellular role are known for one protein, the approach can assign both properties to the novel protein. Both proteins do not have to be evolutionarily related. Approach V: Conserved neighborhood, divergently transcribed genes. Not only gene neighbors that are located in operons are likely to functionally interact: Approach V uses the fact that also neighboring genes that point into diverging, opposite directions frequently encode functionally associated proteins in prokaryotes, especially if this gene organization has been conserved in evolution [11]. Often, one gene within such a so-called ‘divergently transcribed gene pair’ regulates the transcription of the other gene, as well as its own biosynthesis through ‘auto-regulation’. Approach VI: Phyletic vs. phenotypic pattern. The common presence and absence of genes and phenotypic characteristics across species can be used to predict the genes involved in a the manifestation of a phenotype [12-14]. We have developed an novel approach that systematically connects genes and phenotypes by combining genomic context analysis with automated data-mining [15]. In particular, an automated literature analysis searches for documented phenotypic similarities between species. A phenotypic characteristic is then connected to genes that show a presence and absence across species similar to that characteristic.
Figure 1 — Protein function prediction from the genomic context of genes.
References [1] Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., S25:3389-402. [Abstract] [2] Dandekar T, Snel B, Huynen M, Bork P (1998). Conservation of gene order: a fingerprint of proteins that physically interact. Trends Biochem. Sci., 23:324-8. [Abstract] [3] Overbeek R, Fonstein M, D'Souza M, Pusch GD, Maltsev N (1999). The use of gene clusters to infer functional coupling. Proc. Natl. Acad. Sci. U S A, 96:2896-901. [Abstract] [4] Enright AJ, Iliopoulos I, Kyrpides NC, Ouzounis CA (1999). Protein interaction maps for complete genomes based on gene fusion events. Nature, 402:86-90. [Abstract] [5] Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D (1999). Detecting protein function and protein-protein interactions from genome sequences. Science, 285:751-3. [Abstract] [6] Huynen MA, Bork P. (1998). Measuring genome evolution. Proc. Natl. Acad. Sci. U S A, 95:5849-56. [Abstract] [7] Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO (1999). Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc. Natl. Acad. Sci. U S A, 96:4285-8. [Abstract] [8] Galperin MY, Koonin EV (2000). Who's your ne ighbor? New computational approaches for functional genomics. Nat. Biotechnol., 18:609-13. [Abstract] [9] Myllykallio H, Lipowski G, Leduc D, Filee J, Forterre P, Liebl U. (2002). An alternative flavin-dependent mechanism for thymidylate synthesis. Science, 297:105-7. [Abstract] [10] Morett E, Korbel JO, Rajan E, Saab-Rincon G, Olvera L, Olvera M, Schmidt S, Snel B & Bork P (2003). Systematic discovery of analogous enzymes in thiamin biosynthesis. Nat. Biotechnol., 21:790-5. [PDF] [11] Korbel JO, Jensen LJ, von Mering C, Bork P (2004). Analysis of genomic context: prediction of functional associations from conserved bidirectionally transcribed gene pairs. Nat. Biotechnol., 22:911-7. [PDF] [12] Levesque M, Shasha D, Kim W, Surette MG, Benfey PN (2003) Trait-to-gene: a computational method for predicting the function of uncharacterized genes. Curr. Biol. 13:129-33. [Abstract] [13] Makarova KS, Wolf YI, Koonin EV (2003). Potential genomic determbnants of hyperthermophily. Trends Genet., 19:172-6. [Abstract] [14] Jim K, Parmar K, Singh M, Tavazoie S. (2004). A cross-genomic approach for systematic mapping of phenotypic traits to genes. Genome Res., 14:109-15. [Abstract] [15] Korbel JO, Doerks T, Jensen LJ, Perez-Iratxeta C, Kaczanowski S, Hooper S, Andrade M, Bork P (2005). Systematic Association of Genes to Phenotypes by Genome and Literature Mining. PLoS Biol., 3:e134. [PDF]
|
|
Utilizing the genomic context of genes |
